-

航天检测技术(深圳)有限公司
主营:3C认证,CTA入网许可证,交通部794/808认证

航天检测技术(深圳)有限公司
主营:3C认证,CTA入网许可证,交通部794/808认证 8
8

对于新版标准基本规则的一个补充说明:
1) 明确了新版发布前,已评价结束的医疗器械不需要增加新的生物相容性检验;
2)按照新版标准,任何生物相容性检验项目的豁免都需要提供合理解释;
3)历史临床资料可作为符合生物相容性要求的证据以豁**验;
4)任何涉及生物相容性再评价的医疗器械,必须按照新版标准进行再评价。
恩欣格提供的生物相容性声明包括大量必要的数据,有效简化了医疗器械的评估过程。
TECAPEEK MT natural
TECAPEEK MT CF30 black
TECASON P MT blue
TECAFORM AH MT blue
TECAPRO MT white
TECATEC MT PEEK CW50 black
吻合器使用中出现的重大问题,为所有医疗器械的使用敲响了警钟。对于所有准备上市或正在使用的医疗器械,都需要加强以下几个方面的管理,以减少不良事件的发生:卖点切入来挖掘主要是从项目产品出发进行布局保护。例如,戴森的无叶风扇利用了东芝20世纪80年代的过期,从2008年开始布局,2013~2015年发起了20多场诉讼,胜诉率达**,收获了大量利润。
除了急性,还有亚急、亚慢、慢性测试。是由于与人体接触时间更长而需要做的测试。具体对动物进行4周,13周,26周的测试。选择植入,反复注射的方法进行。观察体重、血液生化反应、临床反应、器官重量的变化、病理切片等。这些均是对应高风险类器械的测试。
该声明还确保了可追溯性,这提供了具有成本效益的文档化过程。
药监机构的数据完整性检查缺陷在法规和公众观念方面可能会有很严重的影响,不应该是由于企业缺乏对所需工作的了解而造成。另外,在检查期间,企业没有能力生成所需记录或文件会受到关切,即使不是标准报告或现存质量体系文件,这可能会构成拖延、否决、限制或拒绝检查,这也会产生严重后果。
吻合器虽然方便,但在安全性方面一直存在争议。2017年热播的电视剧《外科风云》中,医生和患者家属就在吻合器的使用问题上产生了分歧,这些问题涉及:医生的使用习惯、医生对于器械的信任程度、手术费用、病人耐受力等。
生物医用材料及其制作与封装的体内植入式器械的生物相容性和相关质量直接关系到患者的生命安全,应该通过严格的生物学评估(biological evaluation),并实行国家统一的注册审批制度,以确保安全。生物学评估可按接触部位(皮肤、粘膜、组织、血液等)、接触方式(直接或间接)、接触时间(暂时、中期和长期)和用途分类,评估的生物学实验项目包括细胞毒性试验、致敏试验、刺激反应试验、亚急性毒性试验、植入试验、血液相容性试验、慢性毒性试验、致癌性试验、生殖与发育毒性试验、生物降解试验等。
测试流程
进行材料的生物学评价过程,第一步是做化学表征测试。比如,医学级材料是否经过USB classic证书,ISO10993测试等。化学测试仅能测量材料本身是否含有有毒有害物质,还需进一步做动物测试来证实安全性,需按照ISO10993-1部分的附录来筛选动物测试,考虑材料在人体内接触的时间和部位,以及临床应用和已有文献数据来确定测试方法和计划。
IVD产品因和人体没有直接接触,不适用ISO10993原则。首先,先看产品与人体有无直接接触,比如膏药,纱布,康复类器械,牙组织体,心血管支架等。间接接触就包括输液器,吸氧雾化器导管,通过管路进入人体内部,经由液体和气体接触人体。如此,判定是否需要按照此流程来进行测试。首先考虑化学表征对人体是安全的。**和无机的化学成分都需要进行测试。通常原材料供应商会提供化学表征测试报告。当采用原材料供应商提供的材料生产医疗器械,也需要提供化学表征测试报告。接下来,考虑材料在上市产品中已有使用。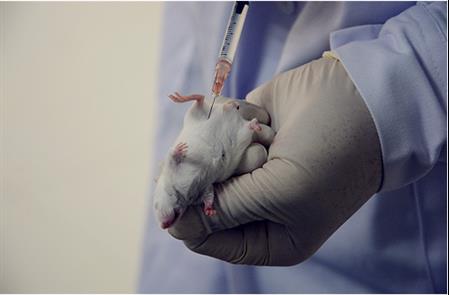
反应测试:新的、动物源性材料可能有学问题,需要考虑。主要包括过敏、抑制、刺激、自身反应等。
ISO10993生物相容性测试是指材料在机体特定部位产生的反应,也就是说某些材料或者药物与人体接触或者植入体内是否能够“相容”,会不会对我们的人体产生伤害。